Copyright © 1998–2025 by A. Miyoshi
SSUMES reference manual - carate/diseig/dislit
SSUMES reference manual
- carate/diseig/dislit/catime/distim
Synopsis
carate basename[.ext]
diseig basename[.ext] [-e]
dislit basename[.ext]
catime basename[.ext]
distim basename[.ext]
Description
The SSUMES component programs
carate/diseig/dislit read in
MASTER input file(s) containing ρ(E) and k(E)
and calculate the steady-state solution to the master-equation for
chemically activated (carate) and unimolecular dissociation and/or
isomerization reactions (diseig & dislit).
The component catime calculates the time-dependent solution to the
chemical activation master-equation, and distim calculates the
time-dependent solution to the dissociation master-equation.
A control input file named basename.inp(.ext)
is used to specify the informations on the wells, names of the MASTER input
files, temperature and pressure, etc.
The results are written to a CSV file named
basename_progname_out.csv.
When the diseig program is invoked with "-e"
option, it also creates a CSV fine named
basename_diseig_eig.csv containing the list of
eigenvalues.
The programs expect following two types of input files usually in
the current directory.
| 1) |
A control file: basename.inp(.ext) |
| 2) |
MASTER file(s) |
The format of the MASTER file is described in the
reference manual for
mas55c3 and original manual for UNIMOL suite.
The structure and allowed keywords in the control file is described in detail
in this manual, and different behavior of the SSUMES program from
mas55c3
in reading MASTER input file(s) is described here.
Control input – structure
The structure of the control input is shown below.
# comments
key value [value2, ...]
well{ ! comments
key value [value2]
}
well{
:
}
At least, one well-block enclosed by
'well{' and
'}' describing the information of a well must present.
The well-block must contain a
'filename' key
specifying the MASTER file name for the well.
For the chemically activated reaction solved by
carate,
One and not more than one well should contain a
'recombChan' key described below.
Comment
Any charactors after and including '!' or '#'
to the end of the line are ignored as comments. There is no
difference between the comments starting with '!' and
'#'.
Keys valid outside blocks
Temperature specifications
Any number of temperature specifications are allowed outside the well-blocks.
There are three types of epecifications listed below. (unit: K)
tempRange T_start T_end T_step
tempRecipRange numer start end step
tempGauChebGrd T_min T_max nT
tempList T1 T2 T3 ...
T0 T0
The key
'tempRange' sets an equally spaced temperatures
list from
T_start to
T_end,
with step
T_step. The next type with a key
'tempRecipRange' sets a list of temperature equally spaced
in the reciprocal of temperature, that is, they are equally spaced in
the Arrhenius plot. Like the convention used for the abscissa of
the Arrhenius plots, the reciprocal temperature may be specified in
the form of: 1000/
T, 10000/
T, or
etc. by
setting the
numer (numerator) to 1000, 10000, or
etc. For example,
tempRecipRange 10000 5 40 1
sets a list of temperatures corresponding to 10000/
T
= 5, 6, 7, ..., 40.
The key starting with
'tempGauChebGrd' sets the list of temperatures at the
Gauss-Chebyshev grid which is suitable for Chebyshev-polynomial fit
of RRKM rate coefficients. The range of temperature is
specified by
T_min and
T_min,
and
nT is the number of grid points.
For the detailed description of the Chebyshev polynomials and
Gauss-Chebyshev grid, see
Chebyshev-polynomial
fit section.
The last type starting with
'tempList' sets a list of temperatures simply as they
listed after the key. If multiple temperature specifications as given,
the merged list of temperatures is used. For example,
tempRange 300 600 100
tempList 298
sets list of temperatures as: { 298, 300, 400, 500, 600 }.
DEFAULT: If no temperature specification is given, programs read them from
the MASTER file(s) and the merged list of them are used.
The key
'T0' sets initial temperature for
distim
solver, and only valid in the
distim input. Default is 298.15 K.
T0 300
Pressure specifications
Following four types of pressure specifications are allowed.
Default unit is Torr.
pressUnit p_unit
pressRange p_start p_end p_step
pressLog10Range log10p_start log10p_end log10p_step
pressGauChebGrd p_min p_max np
pressList p1 p2 p3 ...
The key
'pressUnit' changes the unit of pressure. Allowed
units are;
'Torr' (dafault),
'atm',
'Pa',
'kPa',
'MPa', and
'bar'.
This key affects the pressure specification after it. So, the pressures
may be specified with different unit within a control file. For
example,
pressUnit atm
pressList 1
pressUnit Torr
pressList 0.1 1 10
set the list of pressures as: { 0.1, 1, 10, 760 }.
The key
'pressRange' sets an equally spaced pressure
list from
p_start to
p_end,
with step
p_step. The next type with a key
'pressLog10Range' sets a list of pressures equally spaced
in logarithmic scale. For example,
pressLog10Range -2 2 1
sets the list as: { 0.01, 0.1, 1, 10, 100 }. The unit is affected by
the last
'pressUnit' key before it.
The key starting with
'pressGauChebGrd' sets the list of pressures at the
Gauss-Chebyshev grid which is suitable for Chebyshev-polynomial fit
of RRKM rate coefficients. The range of pressure is
specified by
p_min and
p_min,
and
np is the number of grid points.
For the detailed description of the Chebyshev polynomials and
Gauss-Chebyshev grid, see
Chebyshev-polynomial
fit section.
The last key
'tempList' sets a list of pressures simply as they listed after
the key. When multiple specifications as given, the merged list of them
is used.
DEFAULT: If no pressure specification is given, programs read them from
the MASTER file(s) and the merged list of them are used.
revPressOrd
This key is only valid in
dislit program. This key inverts
the pressure order of calculation, that is, lower pressure to higher.
This is sometimes useful to get converge the desired solution.
Time specifications
Following four types of time specifications are allowed for
catime
and
distim in order specify the output times.
The unit of time is second and the default time base is 1 (s).
Any number of time specifications are allowed outside the well-blocks.
timeBase t_base
timeRange t_start t_end t_step
timeLog10Range log10t_start log10t_end log10t_step
timeList t1 t2 t3 ...
The key
'timeBase' sets the time base used in the subsequent
inputs. for example,
timeBase 1e-6
timeList 1 2 5
timeBase 1e-3
timeRange 3 9 3
sets the output times to 1, 2, and 5 µs and 3, 6, and 9 ms.
The usages of
'timeRange',
'timeLog10Range',
and
'timeList' are similar to corresponding pressure
specifications.
DEFAULT: equivalent to 'timeLog10Range -6 -1 1'.
Solver controls
err3 err
alphaTdep alpha1000 beta
kEthInt kEmin_internal
kEthOut kEmin_outgoing
topCut ntop
maxCyc cyc
sizeLSDSRW size_fact
The parameter
'err3' is the truncation threshold of the
collisional energy transfer probability to make the mastre-equation
matrix banded.
The default is the 1/1000 of the
ERR3 in the MASTER file.
If a MASTER file is generated by
rrkmth,
ERR3 inside it is
1E-6, and, thus, the default is 1×10
−9.
So it may be different from well to well, though there is no physical
background to do so. It should be noted that this parameters must
be set at reasonably small in order to calculate rate paramters at very
high pressure such as 10
10 atm, which is unrealistic but
often needed to obtain the complete fall-off curves.
However, it should not be too small in order to calculate
the steady-state dissociation rate coefficient at low temperatures.
The key 'alphaTdep' can be used specify a
temperature-dependent equation for the collisional energy transfer
parameter α (alpha).
The α is calculated by the equation,
α = α1000 (T / 1000)β,
with α1000 specified by alpha1000
and β specified by beta.
This key overwrites NALPHA and ALPHAV specified in
the MASTER input file(s) for all the wells.
The beta in this key can be any floating-point number,
while it must be an integer or a half-integer in MASTER input.
The parameter 'kEthInt' and 'kEthOut'
are used to remove out very small microscopic rate constants,
k(E), which sometimes make the solver unstable.
Internal k(E)'s, that is, those for isomerization
between wells, smaller than kEmin_internal are
set to 0 (zero). Similarly, external k(E)'s, that is,
those for dissociation or isomerization to species not included as wells,
smaller than kEmin_outgoing are removed.
The default is 1×10−64 for both
kEmin_internal and kEmin_outgoing.
The parameter 'topCut' specifies the number of
grains at top energy to be eliminated from master-equation.
The dafault is 10, which is
a reasonable value needed to eliminates the problem of assymetric
isomerization
k(E) calculated with different number of grains.
The parameter is usually used to reduce the problem size to speed up the
calculation. Note that the default rrkmth generates rather too
much number of grains to avoid the problems at high-temperature, but it
is not always the best choice.
The parameter 'maxCyc' specifies the maximum number of
iteration in dislit program. Other program never read or
use this. The dafault is 300.
The key 'sizeLSDSRW' specifies the factor to the
size of working array for DLSODES routine used in catime and
distim programs.
The default is 0.3. Usually there is no need to adjust this
parameter unless the DLSODES error on the RWORK size is encountered.
Condition control
condition cond
The key
'condition' is used to specify the condition for
the time-dependent solution calculated by
catime (only).
The value
'const' (default) invokes the calculation for
the constant flux condition;
kin =
const.
and
n(
t = 0) =
0, for the master equation,
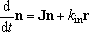 | (10) |
The value
'init' invokes the calculation for the initial
value problem;
kin = 0 and
n(
t = 0) =
r.
Output controls
output opt1 [opt2 ...]
verbose lev
The key
'output' is used to specify the optional outputs.
Allowed options are
'vector',
'HPL',
and
'HPLonly'.
The
'vector' option specifies the output of steady-state
internal population vector, into the file named
basename_progname_vec.csv.
When this option is used for
carate, the incoming-flux vector
is also written in
basename_carate_rfxvec.csv.
The option
'HPL' tells the program to compute high-pressure
limiting (HPL) rate constants by numerical integration and they are written
in a file named
basename_progname_hpl.csv.
When the
'HPL' option is specified together with the
'vector' option for
diseig or
dislit,
the Boltzmann distribution vector is written in
basename_progname_hplvec.csv.
The
'HPLolly' option can be used to compute
HPL rates only.
The options,
'HPL' and
'HPLonly' are allowed
only for
diseig and
dislit.
The key 'verbose' sets the level of the output of
diagnostic messages. The dafault level is 0 and available levels
are 0, 1, 2.
Keys valid inside blocks
Solver/Activation control
recombChan ichan
reactant
excitGrain excitation_grain
excitArbit activation_flux_filename
The key
'recombChan' specifies the recombination channel
number where the reactive flux for chemically activated reaction comes in
for the program
carate. Other programs never read or use it.
ichan is the channel number in the corresponding
MASTER file. Due to the restriction of the
rrkmth program,
the channels are numbered consecutively from that of the lowest to threshold
energy to higher. This key must present in one (and only one) of the
well in the program
carate.
The key
'reactant' (it takes no value) specifies the
reactant well for
dislit in order to solve the steady-state problem
of a type of eq (5) in
introduction to SSUMES.
The program
dislit can be invoked without any
'reactant' key to solve the problem of type of eq (4) in
introduction to SSUMES.
However, the key can be specified in only one well.
New feature in version 2018.06.14
The key 'excitGrain' specifies that the activation flux
comes from a single grain specified by its number for carate and catime.
This correspond to monochromatic photo-excitation followed by internal
conversion which produces highly vibirationally excited ground state molecule.
By the use of 'excitArbit' key, one may specify an arbitrary
initial distribution of a molecule. See activation.csv in
sample folder for the format of the input file. It should be a simple
vector of distribution starting from the ground state of the specified well.
Well properties and attributes
index idx
filename MASTERfile_name
connect idxWell ichan
offset noff
truncate ntrunc
The key
'index' is an optional key to assign indices
to the wells. The well-indices are used in specifying the
connection between wells by
'connect' keyword as well as in
the outputs. The indices should be integer numbers, but not
necessarily the consecutive numbers.
If omitted, indices are automatically assigned
consecutively in the order of appearance in the control input,
starting from 1. However, it is not recommended to use this
default indices in specifying well-connections in many-well problems,
since, for example, when you want to remove well-3 from the 10-well system,
you will need to renumber many indices in many connection specifications.
The name of the MASTER input file corresponding to the well
should to be specified by 'filename' key for each well.
The extension '.dat' is assumed when no extension is given
in the file name. A file with an extension other than
'.dat' may be specified by explicitly specifying the
full file name, but, there is no way to specify the file name without
extension.
The key 'connect' is used to specify the connections
between wells via isomerization reactions. The connection
specifications may appear as many times as required in a well.
The first argument ideWell is the index of the well
to be connected, and the second argument ichan
is the channel number via which the connection is made.
The connections must be consistent in the two-well connected.
The SSUMES programs strictly check the microscopic equilibrium between
the microscopic rate coefficients of the two wells, and the properties
of the transition state connecting between them must be given exactly
the same in the RRKM inputs for two wells.
The 'offset' key is used to tell the programs
about the difference of the energy between vibrationless (ground)
states of isomeric molecules (wells) in the units of grain size,
typically, 100 cm−1.
The value noff must be an integer number.
Since only the differences of noff's among the wells
matter, they may be calculated from any origin, but the origin should
be common among the wells. Usually, the energy of the lowest
well is taken to be the origin, that is, offset 0.
In order to avoid the asymmetry error issued by the SSUMES programs,
it is highly recommended to prepare the RRKM input files with
'alignBaseEnergy' keyword by prepum program
in GPOP.
The 'truncate' key can be used to define the
lower-energy truncation of each well for carate and dislit
solvers in order to include stabilization process. For example,
'truncate 10' means the lowest ten grains of the well
are eliminated from the master-equation. The population flux going
into the lowest ten grains never comes back, and is treated as
stabilized. This key is never read nor used by diseig.
The default is zero (truncate 0) for
all the wells, that is, no stabilization process is included.
Parameters imported from MASTER input file(s)
The SSUMES component programs
carate/
diseig/
dislit
read in MASTER input file(s) compatible for
mas55c3.
Some of the calculation parameters are imported from the MASTER
input file(s) and some of them can be overridden by corresponding
keys in the control input. See the reference manual for
mas55c3
for the detailed description of the MASTER input file. The
different behavior of the SSUMES programs from
mas55c3 are
summarized below.
|
The parameters indicated by
bold Sans-Serif fonts are used by SSUMES
programs. Others are ignored by SSUMES. |
| 1. |
TITLE |
| 2. |
INC,NCHAN,INCCHK |
| |
INCCHK: Read as grain size (cm−1) by SSUMES. |
| 3. |
ERR1,ERR2,ERR3 |
| |
ERR3: tolerance for truncation of P(E,E')
is set to ERR3/1000. |
| 4. |
E0 |
| 5. |
NALPHA |
| |
The number of ALPHAV's read in the next sequence is always 1, irrespective
of this value.
If input as positive (NALPHA > 0) or zero (NALPHA = 0),
constant (templerature independent) α is assumed.
If input as negative (NALPHA < 0), mas55c3 does calculations assuming
the temperature dependent α = α0(T / 1000)β, where α0 is read as ALPHAV(1) and β = |NALPHA| / 2.
For this case, only one value of α is read in the next input. |
| 6. |
ALPHAV |
| |
Value of α or α0 (cm−1). |
| 7. |
IXV,JXV |
| |
The functional form of P(E,E') is always
exponential-down model irrespective of this input in SSUMES. |
| 8. |
NP |
| |
number of pressures. |
| 9. |
PR(I),I=1,NP |
| |
array of pressures, in torr. |
| 10. |
PALMT |
| 11. |
JAV |
| 12. |
NTEMP |
| |
number of input temperatures. |
| 13. |
TEMPV(I),I=1,NTEMP |
| |
array of input temperatures (K). |
| 14. |
SGMA,WT1,WT2,EPS |
| |
See RRKM input. |
| 15. |
IOPTHT,IOPTPR |
| 16. |
NDEGS |
| |
no. of ρ(E) values to be read in. |
| 17. |
RHO(I),I=1,NDEGS |
| |
array of ρ(E) values. |
| 18. |
TEMPV(I),CORRAT(I),I=1,NTEMP |
| 22. |
NRATES |
| |
no. of microscopic rate coefficients k(E) to be input. |
|
- Input no. 25 looped through IN=1,NCHAN |
| 25. |
Rn(I),I=1,NRATES |
| |
list of k(E) for channel n. |
Output
The results are written to a csv-format file,
basename_progname_out.csv.
They are self-instructive. See
Quick-Start for examples and some
instructions.