Fig. Q1
setpath.bat, setpath.csh, and setpath.sh
in the sample folder may be usefull for this purpose.
(Or place the executables in the current directory by copying
/ moving them.)
rc2h2ph_s_rrkm.inp, is an input file
for the rrkmth program, which is a modified version of RRKM program
in UNIMOL. The file contains all necessary information
(vibrational frequencies, rotational constatnts, etc.) on the
(adduct) molecule and transition states for dissociation reactions.
The RRKM input can be prepared manually or by
gpop program suite.
The files used to create rc2h2ph_s_rrkm.inp by prepum
program in gpop can be found in sample/gpop directory.
rc2h2ph_s_mas.dat from an input file
rc2h2ph_s_rrkm.inp. The MASTER input file,
rc2h2ph_s_mas.dat, contains the density of states
and microscopic rate constants for a well necessary for the master-equation
calculations.
rc2h2ph_s_rrkm.inp by
typing:
mas1t.dat, ratnum.csv,
ratnum.dat, ratthm.csv,
rc2h2ph_s_rrkm.out, and tunnel.dat
must have been created in the current directory.
mas1t.dat to rc2h2ph_s_mas.dat
by typing:
move command on recent Windows.)
rc2h2ph_s_ca.inp,
is required to provide the information on the chemical activation reaction
and calculation parameters. Its contents is short and is shown
below.
# phenyl-c2h2 single-well model - chemical activation
tempRecipRange 100000 50 331 20
! 10 atm (default unit of pressure is Torr)
pressList 7600
well{
filename rc2h2ph_s_mas
recombChan 2
truncate 50
}
The lines beginning with '#' or '!' are
comments.
The first two lines except for the comments and blank lines specifies
the temperatures and pressures for calculation.
The block input beginning with 'well{' contains the information
on the well: the name of the MASTER input file containing the density of
states and microscopic rate constants, the channel number where the reactive
flux comes
in ('recombChan'), and the number of the grains to be truncated
('truncate'). This is required to consider the
stabilization process, and the polulation that goes into the lower grains,
for which the number of grains is specified by 'truncate',
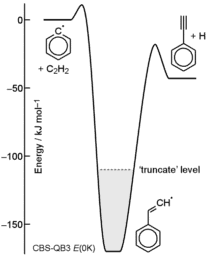
are considered to be stabilized. The level of 'truncate'
is also shown in Fig. Q1.
carate - SSUMES rev. yyyy.mm.dd listWells::alignSize absTop = 792 well-1: err3 = 1e-009 kEthInt / kEthOut = 1e-064 / 1e-064 well-1: recombChan = 2 well-1: truncate = 50 umolProb::initMW psiz = 742 T = 2000 [K], p = 7600 [Torr]: T = 1428.57 [K], p = 7600 [Torr]: : T = 303.03 [K], p = 7600 [Torr]:
rc2h2ph_s_ca_carate_out.csv is created in the current
directory. It contains the essential results of calculations.
Part of the rc2h2ph_s_ca_carate_out.csv is shown below.
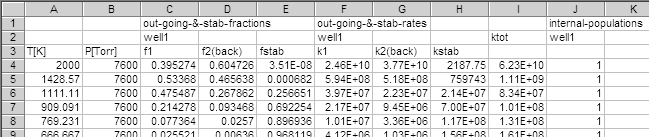
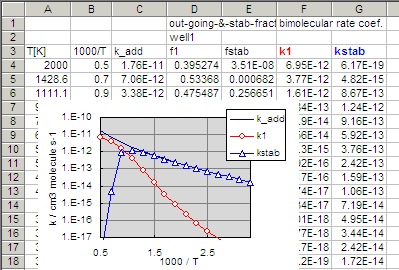
f1 (branching fraction to channel-1),
f2(back) (channel-2), and fstab
(stablization).
Due to the input regulation of rrkmth, the channels are always
numbered consecutively from that with the lowest threshold energy,
E0,
to higher ones. So, the channel-1 in this case is the dissociation
to phenylacetylene + H and channel-2 is the dissociation to phenyl +
acetylene, that is, back dissociation to the reactants (thus it is
indicated as f2(back)).
It should be noted that the rate coefficients in this
output, k1, k2(back), kstab,
and ktot are unimolecular rate constants for the
intermediate in steady-state internal energy distribution, and they
are NOT the bimolecular rate coefficients for phenyl +
C2H2.
f1 and fstab. The high-pressure limiting
rate coefficients must be calculated separately. For example, they
can be calculated by tstrate program in
gpop. Example files for tstrate and results
(r_add.csv) can be found in sample/gpop directory.
An example of the calculation of the bimolecular rate coefficients
is shown below.