basename.csv. An example output is shown below.
Synopsis
Description
vtst reads a VTST reaction input file,
basename.rxv, and GPOP-format files for molecules
or atoms described in the VTST reaction input.
Multiple GPOP-format files created from the frequency jobs along the
reaction coordinate are read in, and the canonical VTST rate constants
are evaluated by interpolation.
Calculated rate constants and equilibrium constants
are written in a CSV file, basename.csv.
gpop1scf reference manual. However, the imaginary frequency
for the tunneling correction, it is used UNSCALED.
Input
| 1) | A VTST reaction input file, basename.rxv | |
| 2) | GPOP-format files (*.gpo) | |
Note: All of the .gpo files should be pre-processed by
gpop3tst before using them with vtst.
Even in the case that no modification is needed, it must be
pre-processed with null .mod files.
The energetics must be specified explicitly in the .rxv
file or by the energyTST in the .mod file
before the processing by gpop3tst.
| ||
basename.rxv, is similar to a .rxn
file for tstrate. It should contain a reactants
block and at least two varTS lines.
Optionally, a products block may be placed.
The following is an example input for the C–H bond
fission reaction of 1H-benzocylobutadiene. The calculation is done
in the reverse direction (recombination).
energyUnit kJ/mol
reactants{
file hatom500 bzcycb2en500
energy 0
}
products{
file bzcycb2en1H500
energy -252.224
}
varTS 1.8 bzcycb2en1Dr018 -54.708
varTS 1.9 bzcycb2en1Dr019 -31.200
varTS 2.0 bzcycb2en1Dr020 -15.864
varTS 2.1 bzcycb2en1Dr021 -7.521
varTS 2.2 bzcycb2en1Dr022 -3.386
varTS 2.3 bzcycb2en1Dr023 -1.407
varTS 2.4 bzcycb2en1Dr024 -0.502
varTS 2.5 bzcycb2en1Dr025 -0.124
varTS 2.6 bzcycb2en1Dr026 0.017
varTS 2.7 bzcycb2en1Dr027 0.081
varTS 2.8 bzcycb2en1Dr028 0.214
The reactants or products block should contain at least one
file key, specifying the name of '.gpo'
file(s) to be read.
Unlike the .rxn file, the file name and energy along the
reaction coordinates are specified by a line beginning with
varTS by the following format.varTS coord filename [energy]Here,
coord is the reaction coordinate in arbitrary
unit, filename is the base name of a .gpo
file, and energy is the zero-point energy adiabatic
energy at this position in the unit specified by energyUnit.
If the energy to be used is already set in each .gpo
file using energyTST by gpop3tst, the last
parameter may be omitted.
The varTS should appear in the increasing order of the coordinate.
All valid keys in the blocks or the outside the blocks will be described
in detail below.
energyUnit unit
energy key in the blocks.
Note that this NEVER override the unit of energyTST key
in '.gpo' files, which is always hartree.
Values may be one of 'hartree' (default),
'kJ/mol', and 'cm-1'.
tempRange T_start T_end T_step tempRecipRange numer start end step tempGauChebGrd T_min T_max nT tempList T1 T2 T3 ...
tempRange or
tempList can be used. Same as these keys in the
temperature file format in
gpop4thf.
fileType ftyp
gpo' (default) and 'tst'. This is retained for
older compatibility.
file filename1 [filename2 ...]
.gpo).
energy engy
energyUnit' key outside the block.
Output
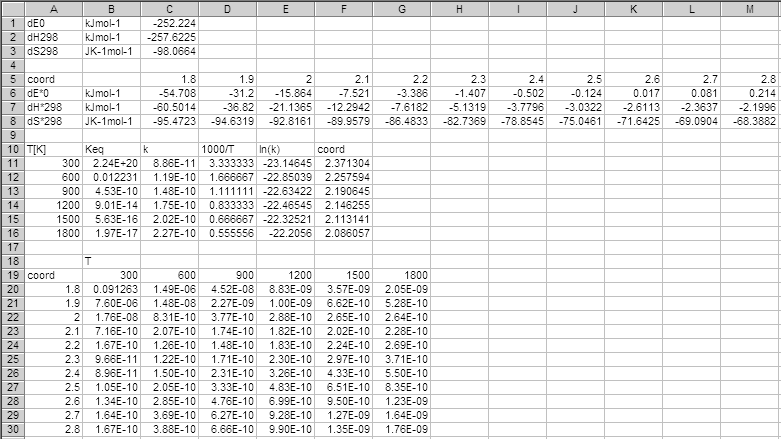
basename.csv. An example output is shown below.
.rxv file, and show the internal energy change at 0 K (dE0),
enthalpy change at 298.15 K (dH298), and entropy change at 298.15 K (dS298)
of the reaction. The fifth to eighth rows show the activation
internal energy at 0 K (dE*0), activation enthalpy at 298.15 K (dH*298),
and activation entropy at 298.15 K (dS*298) at each coordinate.
It is recommended to check these values, especiallly when you are
to use the energy already set in the .gpo files.
vtst program searches for the minimum ln(k) along
the reaction coordinate by the third-order natural spline interpolation.
The coord output at each temperature must be carefully
checked for not being the either edge of the input coordinates, which
suggests the true minimum will be outside the input range of the coordinate.
The last output block contains the rate constant calculated at each
coordinate. This is also helpful to check whether the minimum is
located in the input range of the coordinate.