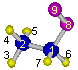Copyright © 2002–2025 by A. Miyoshi
GPOP - Quick Start step-1
GPOP - Quick Start step-1
Followings are examples of the usage of GPOP tools for examination
of intramolecular rotation etc.
Preparation
- Build GPOP from source files or unpack pre-built binaries for Windows
according to the instructions in
Installation section.
- Set PATH to the directory where GPOP executables reside.
The
setpath.bat, setpath.csh, and setpath.sh
in the sample folder may be usefull for this purpose.
Then, change directory to the gpop/sample.
- Alternatively, if you found any difficulty in setting PATH,
copy all GPOP executables (in
gpop-winXX-binary) and
all sample files (in gpop/sample) to the current directory.
Pre-processing
- Execute
gpop1scf for etp500.log by
typing:
gpop1scf etp500
- Files
etp500.gpo and etp500.mod
(or etp500_.mod)
must have been created in the current directory.
- The file,
etp500.gpo, is a GPOP
format file containing essential results extracted from
etp500.log.
A GPOP format file is an ASCII text file and may be viewed with
text editors. This file only may be usefull for some purposes:
for example, the value of SCFenergy key in
jobInfos block is the final SCF/DFT energy at optimized
geometry.
Calculation of properties of an intramolecular rotation
(Skip this part if you do not like to learn about the intramolecular rotation right now.)
- Execute
gpop6irt as;
gpop6irt etp500 8-9 1-@
- Calculation is made for intramolecular rotation around
C[1]–O[8] bond of the ethyl peroxy radical shown below.
- The properties of the intramolecular rotation specified in the
command line are printed to the standard output as follows:
base file name: etp500
moiety-1: 8-9
moiety-2: 1-2-3-4-5-6-7
two moieties are whole molecule.
dihedral angle between moieties 1 and 2: 71.4081
[moiety-1]
appI[amuA2]: 24.05850766
appB[cm-1]: 0.70069305
--- symmetric top ---
symRedI[amuA2]: 6.66957233
symRedB[cm-1]: 2.52754274
--- asymmetric top ---
asmRedI[amuA2]: 7.46994390
asmRedB[cm-1]: 2.25672767
[moiety-2]
appI[amuA2]: 37.50552344
appB[cm-1]: 0.44947057
--- symmetric top ---
symRedI[amuA2]: -4.75414496
symRedB[cm-1]: -3.54588033
--- asymmetric top ---
asmRedI[amuA2]: 7.46994389
asmRedB[cm-1]: 2.25672767
[most resembling vibrations]
mode-001: 0.99820037
mode-010: 0.53138804
mode-004: 0.46718380
mode-009: 0.29262286
mode-020: 0.24449702
- In most cases, the required results can be found in the last
of
[moiety-1] section. The reduced moment of inertia
for rotation around C–O bond is 7.470 amu Å2,
or the corresponding rotational constant is 2.257
cm–1. Also the last [most resembling
vibrations] section indicate that the rotation around
C–O bond well resembles the vibrational mode #1.